LammpsでNVEアンサンブルで計算する際に体積を変化させたい。
change_boxコマンド
change_boxで可能。
https://docs.lammps.org/change_box.html ⧉
とりあえずmeltでやってみる。
# 3d Lennard-Jones melt
units ljatom_style atomic
lattice fcc 0.8442region box block 0 10 0 10 0 10create_box 1 boxcreate_atoms 1 boxmass 1 1.0
pair_style lj/cut 2.5pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 binneigh_modify every 20 delay 0 check no
change_box all x scale 2.0 y scale 2.0 z scale 2.0
velocity all create 3.0 87287 loop geom
fix 1 all nve
dump 1 all atom 25 *.dumpdump_modify 1 pad 4
thermo 50thermo_style custom step temp press pe ke etotalrun 10000‘change_box’だけだと、周りに空間ができるだけで、原子間の構造・距離は変わらない。
シミュレーションセルの変更に合わせるように原子位置も変えたい場合はremapをつける。
# 3d Lennard-Jones melt
units ljatom_style atomic
lattice fcc 0.8442region box block 0 10 0 10 0 10create_box 1 boxcreate_atoms 1 boxmass 1 1.0
pair_style lj/cut 2.5pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 binneigh_modify every 20 delay 0 check no
change_box all x scale 2.0 y scale 2.0 z scale 2.0 remap
velocity all create 3.0 87287 loop geom
fix 1 all nve
dump 1 all atom 25 *.dumpdump_modify 1 pad 4
thermo 50thermo_style custom step temp press pe ke etotalrun 10000lammpsのコマンドで構造を作る場合、格子定数を変えれば対応できるのであまりありがたみは感じないが、read_dataで構造を読み込んで使う場合は便利だと思う。構造ファイルをいじるのはめんどくさい。
おまけの考察
上の計算において、remapをつけようがつけまいが最終的にはどちらも同じ状態に近づくと思っていたが、そうではなかった。
remapをつけた計算では、原子がランダムに飛び回る均質な組織になっているが、remapをつけない計算では、ある程度時間が経過したのち原子のクラスタリングが見られる(ダマになって
いる)。
log.lammpsから、エネルギーを取り出してプロットしたのが以下。
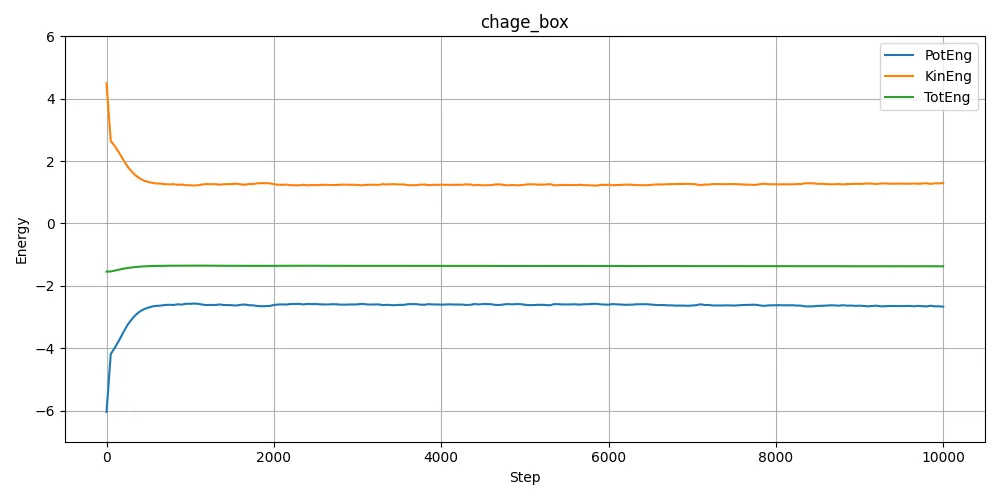
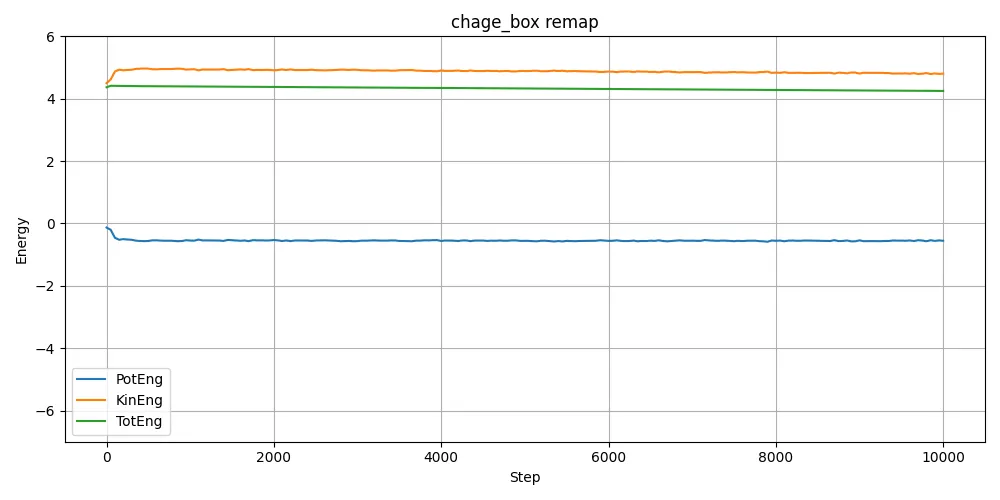
計算スタート時の構造(原子間距離)が違うことでポテンシャルエネルギーが違うため、当たり前といえば当たり前なのだが、計算してみるまで当たり前のことも見落とすことも多い。もっと大きな運動エネルギーを与えれば最終的に行き着く組織は同じになるだろうとは思うが。与えた温度が絶妙なのだろう。
