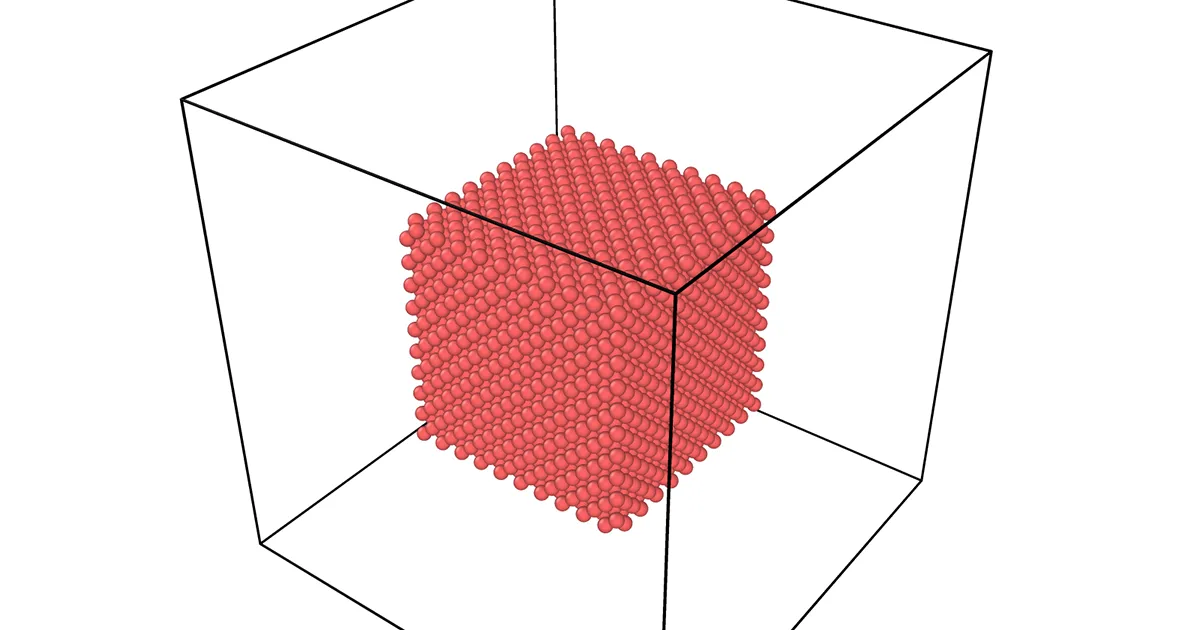
When performing simulations under the NVE ensemble in LAMMPS, I wanted to find a way to change the simulation cell volume during the run.
change_box Command
This can be achieved using the change_box command.
https://docs.lammps.org/change_box.html ⧉
As a test case, I tried this using the melt example.
# 3d Lennard-Jones melt
units ljatom_style atomic
lattice fcc 0.8442region box block 0 10 0 10 0 10create_box 1 boxcreate_atoms 1 boxmass 1 1.0
pair_style lj/cut 2.5pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 binneigh_modify every 20 delay 0 check no
change_box all x scale 2.0 y scale 2.0 z scale 2.0
velocity all create 3.0 87287 loop geom
fix 1 all nve
dump 1 all atom 25 *.dumpdump_modify 1 pad 4
thermo 50thermo_style custom step temp press pe ke etotalrun 10000Using change_box by itself only changes the simulation box size — it just adds empty space around the atoms, and their positions/distances remain unchanged.
If you want the atomic positions to scale accordingly with the new box dimensions, add the remap option to the command.
# 3d Lennard-Jones melt
units ljatom_style atomic
lattice fcc 0.8442region box block 0 10 0 10 0 10create_box 1 boxcreate_atoms 1 boxmass 1 1.0
pair_style lj/cut 2.5pair_coeff 1 1 1.0 1.0 2.5
neighbor 0.3 binneigh_modify every 20 delay 0 check no
change_box all x scale 2.0 y scale 2.0 z scale 2.0 remap
velocity all create 3.0 87287 loop geom
fix 1 all nve
dump 1 all atom 25 *.dumpdump_modify 1 pad 4
thermo 50thermo_style custom step temp press pe ke etotalrun 10000If you’re generating structures using LAMMPS lattice commands, it’s usually easier to just change the lattice constant. So in that case, change_box might not feel very useful. However, if you are loading a structure from a read_data command, this becomes quite handy — it saves the trouble of modifying the structure file manually.
Discussion
Initially, I assumed that simulations with or without the remap option would eventually converge to the same atomic configuration — but that wasn’t the case.
In the run with remap, atoms end up evenly distributed and exhibit a homogeneous structure.
In contrast, in the run without remap, atoms begin to form clusters (aggregated “blobs”) after some simulation time.
I extracted energy data from log.lammps and plotted the results.
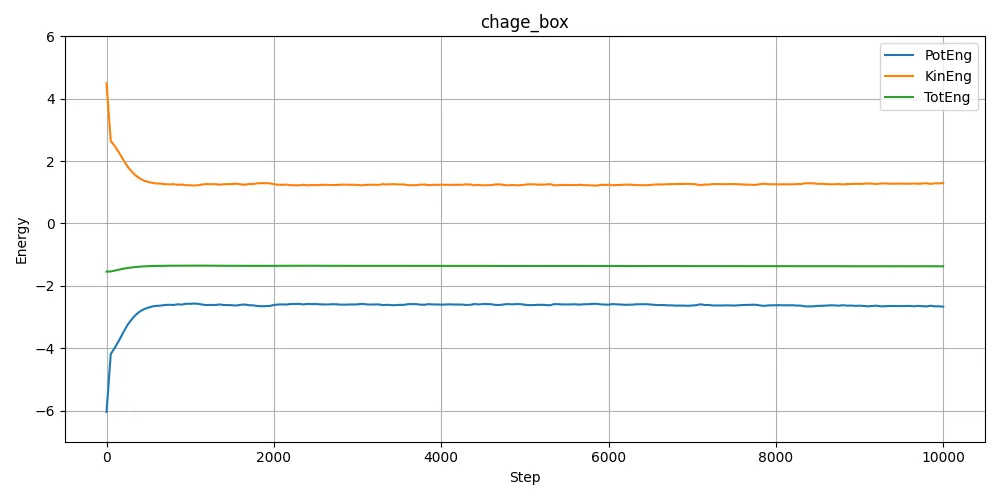
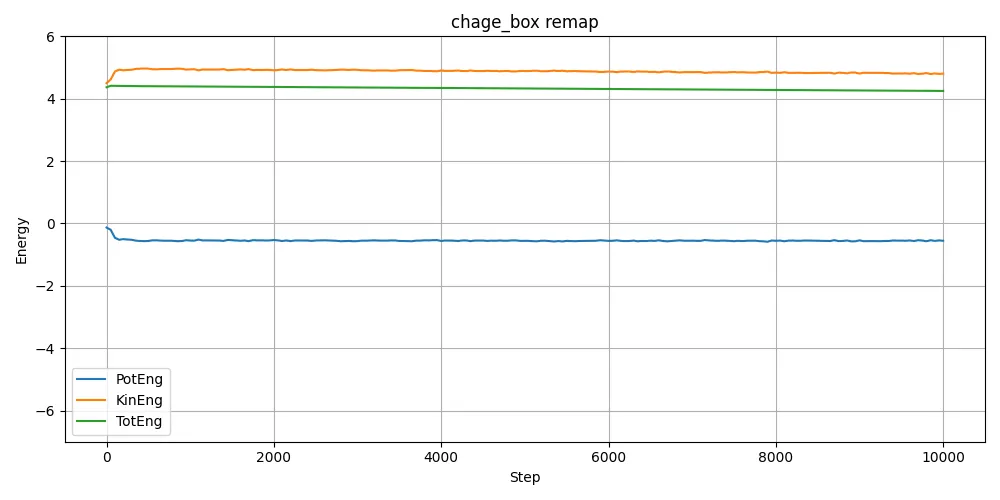
Since the initial atomic arrangements (and hence potential energy) are different depending on whether remap is used, the divergence in behavior makes sense in hindsight. It’s one of those things that seems obvious after seeing the results — but it’s easy to overlook beforehand.
I suspect that if I had given the system more kinetic energy, both cases would eventually converge to the same structure. The difference is likely due to the specific temperature I chose—it was just enough to make the outcome sensitive to the initial conditions.
